
Abstract
Pendula-phenotyped Norway spruce has a potential forestry interest for high-density plantations. This phenotype is believed to be caused by a dominant single mutation. Despite the availability of RAPD markers linked to the trait, the nature of the mutation is yet unknown. We performed a quantitative trait loci (QTL) mapping based on two different progenies of F1 crosses between pendula and normal crowned trees using NGS technologies. Approximately 25% of all gene bearing scaffolds of Picea abies genome assembly v1.0 were mapped to 12 linkage groups and a single QTL, positioned near the center of LG VI, was found in both crosses. The closest probe markers placed on the maps were positioned 0.82 cm and 0.48 cm away from the Pendula marker in two independent pendula-crowned × normal-crowned wild-type crosses, respectively. We have identified genes close to the QTL region with differential mutations on coding regions and discussed their potential role in changing branch architecture.
Similar content being viewed by others


Introduction
The molecular control of lateral branching involves phytohormones such as cytokinins, auxin (IAA), and strigolactones (Leyser 2008). Even though plant architecture-related pathways are fairly well understood in model species (Jiao et al. 2021; Roychoudhry and Kepinski 2015; Sakai and Haga 2012; Strohm et al. 2013), the genetic regulation of branching architecture in trees, and especially in conifers, is overall poorly investigated. During the last decade, several studies have been conducted on branching mutants in different angiosperm tree species in order to identify the responsible genes underlying the branching phenotype. The TAC1 gene is responsible for vertically oriented growth of branches and mutations in TAC1 have been shown to result in “pillar” phenotypes in peach (Prunus persica) (Dardick et al. 2013), plum (Prunus domestica) (Hollender et al. 2018b), Populus × zhaiguanheibaiyang (Xu et al. 2017), and black cottonwood (Populus trichocarpa) (Fladung 2021). Similarly, LAZY1 has been shown to regulate the horizontal orientation of lateral shoots (Xu et al. 2017). Wider branch angles are regulated by WEEP gene in peach, causing a more pendulous phenotype (Hollender et al. 2018a).
Trees with narrower crowns, either caused by very small or very large branch angles, can potentially be planted closer together and thereby give a possibility to utilize the planting area more efficiently. In Norway spruce (Picea abies L Karst.), a branching mutant entitled “Pendula” has been identified and this mutation is characterized by down-oriented lateral branches (Fig. 1). Earlier studies have shown that the harvest index of Pendula individuals is higher than wild-type trees, especially the above-ground one (Pöykkö and Pulkkinen 1990; Pulkkinen and Poykko 1990). They also appear to be less sensitive to tree competition than normal-crowned individuals (Gerendiain et al. 2008), which suggests that they can be planted in denser stands. This mutant also appears to segregate in a 1:1 dominant segregation pattern consistent with the control of one or only a few genes with no intermediate phenotype (Karki and Tigerstedt 1985; Lepistö, 1985). However, in order to utilize the Pendula mutant in practical forestry and silviculture, a method for screening the branch phenotype at an early age is necessary. This could be done with a reliable genetic marker. An earlier study aimed at identifying such a marker was conducted by Lehner et al. (1995) using random amplified polymorphic DNA (RAPD) markers to map the Pendula gene using bulked segregant analysis in 43 full-sib progenies from the cross P289 (normal crowned) × E477 (Pendula). One marker, OPH10_720, was found to be linked to the Pendula gene with an estimated recombination frequency of 0.046 (SE = 0.032) (Lehner et al. 1995), but RAPD markers are known to have low reproducibility, making them unsuitable for large-scale screenings.

Pendula (left) and regular (right) Norway spruce trees. Photo taken at Arboretum Norr in Umeå, Sweden. Photo taken by Carolina Bernhardsson, summer 2013
The advent of next-generation sequencing (NGS) technologies and the availability of a draft genome assembly for Norway spruce (Nystedt et al. 2013) have opened up possibilities to explore the genetic architecture underlying the Pendula phenotype by creating dense genetic maps and subsequent quantitative trait loci (QTL) mapping of the phenotype. Here, we present four genetic maps, two maternal and two paternal genetic maps, derived from QTL mapping in two independent F1 crosses of a Pendula and normal crowned parent (E477 × K954 and E479 × E2089, respectively).
Materials and methods
DNA extraction and sequence capture
Newly flushed buds were collected from two progeny trials of F1 crosses between a Pendula and a normal crowned individual made in the 1980s (E477 (pendula) × K594 and E479 (pendula) × E2089) at a breeding trial run by the Natural Resources Institute Finland (LUKE, formerly METLA) in the spring/summer of 2013. At the same time, branching phenotypes were documented in all offsprings, resulting in 418 and 371 collected and documented progenies for cross E477 × K594 and E479 × E2089, respectively. The samples were shipped to Umeå University, Sweden, for DNA extraction.
DNA extraction was performed using a Qiagen DNeasy® Plant Mini Kit with approximately 20 ng of freeze-dried tissue as starting material and using the default protocol. Each extracted sample was measured for DNA quality using a Qubit® ds DNA Broad Range (BR) Assay Kit, and all samples had DNA concentrations of ≥ 21.4 ng/µl (mean 66.2 ng/µl) and a total amount of DNA ≥ 2.2 µg (mean 7.4 µg). The 828 samples, including samples of the four parents, were sent to RAPiD Genomics© (Gainesville, FL, USA) in October 2014 for sequence capture using 40,018 capture probes that had been specifically designed to target 26,219 partially validated gene models from the P. abies genome assembly (Vidalis et al. 2018). Where possible, probes were designed to flank regions of known contig joins in the v1.0 genome assembly of P. abies (Nystedt et al. 2013, for further detail on the probe design, see Vidalis et al. 2018).
SNP calling and filtering and map creation
The capture data was sequenced by RAPiD Genomics© on an Illumina HiSeq 2000 in either 2 × 125 or 2 × 75 bp sequencing mode with a mean and median of 14 × and 15 × , respectively, of sequencing depth in October 2015. Due to the low sequencing depth of the parental samples, these were re-sent for a second round of sequence capture to increase the genotyping call rate of the parents. The raw sequencing reads were mapped against the complete P. abies reference genome v.1.0 using BWA-MEM v.0.7.12 (H. Li and Durbin 2009). The two BAM files for each parental sample (caused by the two rounds of sequencing) were merged using Samtools v.1.2 before further processing through the variant calling pipeline. Following read mapping, the BAM files were subsetted to only contain the probe-bearing scaffolds (a total of 24,920 scaffolds) using Samtools v.1.2 (H. Li et al. 2009; H. Li and Durbin 2009). Duplicates were marked and local realignment around indels was performed using Picard (http://broadinstitute.github.io/picard/) and GATK (https://software.broadinstitute.org/gatk/) (DePristo et al. 2011; McKenna et al. 2010). Genotyping was performed using GATK Haplotypecaller (version 3.4–46) (DePristo et al. 2011; Van der Auwera et al. 2013), with a diploid ploidy setting and GVCF output format. Combined GVCF was then run on batches of ~ 200 GVCF to hierarchically merge them into a single GVCF and a final SNP call was performed using genotype GVCF jointly on the 5 combined GVCF files, using default read mapping filters, a standard minimum confidence threshold for emitting (stand-emit-conf) of 10, and a standard minimum confidence threshold for calling (stand_call_conf) of 20. See Vidalis et al. (2018) for a full description of the pipeline used for calling variants.
The raw VCF-file including the 828 samples was split into each of the crosses separately so that the VCF-file for the E477 × K954 cross-contained 435 progenies plus parents, and the VCF-file for the E479 × E2089 cross-contained 389 progenies plus parents. The two VCF-files were then filtered so that only bi-allelic SNPs within the extended probe regions (120 ± 100 bp) and without any low-quality tags (QUAL < 20) were kept. To increase the chance of capturing the true genotypes, per site sample genotypes were recoded to missing data if they had < 5 × coverage or a genotype quality < 10. Principal component analyses (PCAs) were performed on the relatedness estimates from vcftools v. 0.1.12b –relatedness (Danecek et al. 2011), and mislabeled progenies, i.e., progenies not related to both parents, were removed (Supplementary Figure S1). The last pruning step was conducted by removing all SNPs showing > 50% missing calls, resulting in a final data set containing 376 samples (including parents) and 333,859 SNPs for the E477 × K954 cross, and 346 samples (including parents) and 317,071 SNPs for the E479 × E2089 cross.
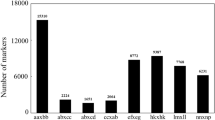
The genotype data were then exported from the VCF-files and all remaining analyses were conducted with R (R core team 2013). The data sets were thereafter further filtered so that only SNPs where at least one of the parents was heterozygous were kept. Progeny genotype calls were then recoded to missing data if they showed genotypes that were not included in a Punnet square based on parental genotypes, and progenies with > 50% missing calls and SNPs with > 20% missing data were filtered out. Due to the risk of collapsed regions caused by the complexity of the reference genome, a test for segregation distortion (Mendellian violations) was conducted on the remaining SNPs using a chi-square test, and all SNPs with a p-value > 0.005 were kept and considered as informative markers. Each of the informative markers got assigned to the probe region they belonged to, and for each probe, only the most informative marker in terms of the least amount of missing data and most balanced segregation pattern was kept for map creation. To avoid saturating the mapping algorithm, the marker with highest genotype frequency was kept and then, if a tie, kept the marker with most balanced segregation according to parental genotypes. Due to the LD between markers, true associations to other positions would still be captured in filtered data. Finally, the Pendula phenotype was included as a pseudo-genetic marker (Pendula marker) and the data were recoded into BatchMap input format (Schiffthaler et al. 2017). This resulted in 340 F1 progenies (175 pendula and 165 normal crowned) and 9737 markers for cross E477 × K954 and 306 F1 progenies (127 pendula and 179 normal crowned) and 16,687 markers for cross E479 × E2089. Differences in the number of markers found between crosses are due to differences in DNA quality/sequencing success of the samples.
Framework genetic maps were then created separately for the two crosses using BatchMap (Schiffthaler et al. 2017), a parallelized version of OneMap (Margarido et al. 2007), using a pseudo test cross strategy. To reduce the number of redundant markers in the map, identical markers (showing no recombination events between them) were grouped into bins and one marker from each bin was used as a bin representative in the map creation. Pairwise estimates of recombination frequencies were calculated between all marker pairs using a LOD score of 14 and a maximum recombination fraction (max.rf) of 0.35. Markers were grouped into linkage groups (LG) and split into maternal and paternal testcrosses. Each testcross LG was ordered with the “record.parallel()” function using 20 ordering runs over 20 CPU cores. The genetic distance between ordered markers was calculated using the “map.overlapping.batches()” approach with 25 markers overlap between batches, a batch size of ~ 50 and a ripple window of 11, all parallelized over 2 phase CPU cores and 20 ripple CPU cores. Highly probable mis-ordered markers, i.e., markers showing a recombination fraction distance to the closest neighbor on both sides of > 0.05, were removed and the full testcross LG was recalculated with a new run of “map.overlapping.batches()” and the same settings as previously. Finally, to minimize the effect of genotyping errors on map size, we counted the number of double recombination events in sliding windows of three markers along the testcross LG and thereafter corrected the genetic distances accordingly as suggested by Cartwright et al. (2007).
To anchor the two crosses LG and give them a common name, the number of shared scaffolds between each LG and a previous haploid consensus map for P. abies (Bernhardsson et al. 2019) was used. All LG were renamed to the haploid consensus map LG names (Supplementary Figure S2).
QTL mapping and search for candidate genes
Associations between all markers placed on the genetic maps and the branching phenotype of progenies (pendula or normal crowned) were tested with chi-square tests. The − log10 (p-value) of the associations were then plotted against the marker position on the testcross to identify the position(s) of any QTL. To compensate for the lower p-values of double heterozygous markers (segregating in both parents), caused by an extra degree of freedom in the analyses, the p-values for these associations were multiplied by two in the female maps (E477 and E479).
All scaffolds showing marker associations of − log10 (p-value) > 40 were considered as candidates for harboring the Pendula locus and all gene models from these scaffolds were extracted and evaluated in Congenie (http://congenie.org) for their gene ontology (GO) and protein family (PFAM) descriptions and compared to the Arabidopsis thaliana (Arabidopsis) database at http://atgenie.org. Each transcript sequence was also compared to the NCBI blastp database (https://blast.ncbi.nlm.nih.gov) to further analyze the function of the genes (Supplementary File 1).
Candidate genes known to be responsible for branching architecture phenotypes in different angiosperms were positioned on the maps by extracting their corresponding putative conifer sequence ID. This data was obtained either from earlier published articles or, when unknown, by performing the BLAST with known gene model sequences against the Norway spruce draft assembly using blastx at http://congenie.org. The same procedure was performed for genes known to be part of the gravitropism and phototropism pathways in plants (reviewed in Bemer et al. 2017; Hollender et al. 2018b; Hollender and Dardick 2015; Jiao et al. 2021; Roychoudhry and Kepinski 2015; Sakai and Haga 2012; Strohm et al. 2013) by searching for the genes at atgenie.org and thereafter identifying the corresponding gene models in P. abies that belong to the same orthologous gene family and if possible place them on the genetic maps based on scaffold position (Supplementary File 2).
The best match or ortholog for the Norway spruce genes with non-synonymous SNPs was detected in Arabidopsis by performing Blastp in PlantGenIE (https://plantgenie.org), TAIR (https://www.arabidopsis.org/index.jsp), and NCBI (https://www.ncbi.nlm.nih.gov/). The domain regions of these genes (Supplementary Figures S3–S8) were confirmed by referring to its best match in Arabidopsis thaliana and by performing searches in the Conserved Domain Database (CDD) (Marchler-Bauer et al. 2015), UniProt (Bateman et al. 2021), and referring to the literature (Pang et al. 2014; Wagner et al. 2002).
Results and discussion
Two F1 crosses were used in the QTL mapping of the Pendula phenotype by creating two independent sets of parental genetic linkage maps. These maps were then used to position associations with the qualitative phenotype (pendula or wild-type) using chi-square tests. A total of 19,139 markers from 14,997 gene-bearing scaffolds of the Norway spruce draft assembly v.1.0 (Nystedt et al. 2013) could be placed on the maps. This corresponds to 25.4% of all scaffolds harboring annotated gene models in the v1.0 P. abies assembly (14,997/58,983 scaffolds), which anchors 9070 high confidence (HC), 6802 medium confidence (MC), and 2042 low confidence (LC) gene models to the genetic maps, corresponding to 26.9% of all partially confirmed gene models in the annotation file (17,914/66,632).
The first cross, E477 × K954, contained 340 progenies and 9714 segregating markers, with 5525 markers positioned on the female map (E477) and 5725 markers on the male map (K954). The total size of the parental maps was estimated to 3585 cm and 3571 cm, respectively (Table 1). The second cross, E479 × E2089, contained 306 progenies and 16,658 segregating markers, with 9392 markers positioned on the female map (E479) and 9786 markers on the male map (E2089). The total size of these parental maps was estimated to 3393 cm and 3115 cm, respectively (Table 1). All four parental maps were grouped into 12 LG each, corresponding to the haploid number of chromosomes in P. abies (Sax and Sax 1933).
The number of unique probe markers per scaffold that were anchored to any of the two crosses parental maps ranged between one and six with an average of 1.28 (median 1). 1.4% of all scaffolds (207 out of 14,997) and 5.3% of multi probe marker scaffolds (207 out of 3882) have markers that map to different LG but where the LG grouping is consistent between the two crosses. However, 51 markers (0.27% of all markers and 0.7% of all markers present in both crosses), distributed over 48 different scaffolds, do not show consistent grouping to LG between the two crosses (Supplementary File 3). When comparing the order of shared markers along the LG between parental maps, the estimated correlations (Kendall’s tau) range between 0.97 and 0.99. Ranges are in line with previously estimated correlations between different maps in P. abies (Bernhardsson et al. 2019).
The significance of the associations between marker genotypes and the Pendula phenotype ranged from a − log10 (p-value) of 0 to 55.1 with a mean of 1.0 and a median of 0.38 for cross E477 × K954. For cross E479 × E2089, the − log10 (p-value) ranged from 0 to 60.8 with a mean of 1.2 and a median of 0.34. A single large QTL could be detected in each of the crosses, with a peak positioned close to the center of LG VI (Fig. 2). The Pendula genetic marker that was added to the linkage maps for reference is positioned in the middle of the QTL in both female maps (black horizontal line for E477 and E479 on LGVI in Fig. 2). None of the markers located on any of the other 11 LG shows any evidence of association with the Pendula phenotype. The four parental framework maps show slightly different marker orders (order correlations ranged between 0.97 and 0.99), but the overall genomic patterns are the same (Fig. 3). However, this inconsistency in marker order within shorter regions of a genetic map is quite typical and has been seen in several other previously published maps. This is most probably caused by a combination of the heuristic ordering algorithms used for creating dense maps and possible genotyping errors caused by insufficient sequencing depth at some markers (Kelley and Salzberg 2010; Khan et al. 2012; Salzberg and Yorke 2005). Since the Pendula phenotype behaves like a qualitative rather than quantitative trait, we chose to include the phenotype as a pseudo-genetic marker in the genetic maps in addition to performing associations against all other markers. The peak of the QTL and position of the Pendula trait “marker” falls at the same location in both maps, which strengthens the robustness of the results. All scaffolds with top associations, − log10 (p-value) > 40 for markers segregating only in mothers and − log10 (p-value)*2 > 40 for markers segregating in both parents, were investigated for containing candidate genes. In total, 169 probe markers distributed over 146 scaffolds show high associations. These scaffolds contain 181 annotated gene models of which 131 gene models have orthologous gene family members in Arabidopsis (Supplementary File 1).

Marker order correlations of LG VI between parental maps. Red dot in top right figure shows the position of the pendula marker between E477 and E479. Red dotted lines show the position of the pendula marker when only one of the compared parental maps harbor the marker (E477 or E479). The order correlation, estimated with Kendall’s tau, is shown in the top left corner of each plot

Alignment of the haploid Linkage groups (LG) and significance of the associations between marker genotypes and the Pendula phenotype
Previously identified genes responsible for branching architecture (Bemer et al. 2017; Hollender et al. 2018b; Hollender and Dardick 2015; Jiao et al. 2021; Roychoudhry and Kepinski 2015; Sakai and Haga 2012; Strohm et al. 2013) were aligned to the Norway spruce draft assembly and, if found, anchored to the genetic map. A total of 391 gene models were tested, belonging to 28 gene families associated with gravitropism/phototropism (including WEEP1, WEEP2, LAZY1, TAC1, and FUL). Overall, 118 of these gene models (30.2%), from 21 gene families, could be anchored to the genetic maps and are distributed over all 12 LG and their corresponding putative conifer sequence ID (MA_181281g0010 and MA_10435286g0010 for WEEP, gi|21,187,158, gi|49,450,754 for LAZY1, gi|69,453,051 for TAC1, and no clear sequence for FUL) were identified in the Norway spruce v.1.0 genome assembly. The WEEP genes that have been found to yield a pendulous phenotype in peach (Prunus persica) to Hollender, Waite, et al. (2018) are positioned on LG X (WEEP1, MA_181281g0010) and LG V (WEEP2, MA_10435286g0010). The IGT gene family, which harbors several genes known to influence plant architecture in angiosperms, including LAZY, TAC (tiller angle control), and DRO (deeper rooting) (Hollender et al. 2018b; Jiao et al. 2021; Waite and Dardick 2020), appears to be a single copy gene in P. abies as we can only identify a single homolog, MA_39199g0010. This gene is positioned at the distal ends of LG I. Nineteen of the gene models, from 10 different gene families, are located on LG VI. However, none of the gene models anchored to LG VI is positioned close to the center of the QTL (Supplementary File 2). Even though the list of candidate genes positioned within a 5 cm genetic distance from the Pendula marker is still fairly large (Supplementary Table S1 and S2), we can now rule out most of the earlier known genes involved in tree branching architecture, including the gravitropism and phototropism biosynthesis pathways.
For female E477, 43 probe markers, from 41 different scaffolds, are positioned within a 5 cm distance from the Pendula marker, with the closest markers occurring at a distance of 0.82 cm (Supplementary Table S1). Seven probe markers are positioned at this closest distance and one of them, MA_10436629:1, does also show the most significant association for the whole cross (p-value 5.58e-56). This probe marker is located within the gene model MA_10436629g0010, which transcribes an adenylosuccinate synthase (ADSS) (Supplementary Table S2) and is also present in the cross E479 × E2089, positioned 12.81 cm away from the pendula marker on the E477 map but showing a highly significant association (p-value 9.53e-60). This gene class is involved in the de novo purine biosynthesis pathway (Stayton et al. 1983), which is a central metabolic function (Smith and Atkins 2002) and has been described to express at higher levels in the shoot apex vegetative, young leaves, and rosette (bar.utoronto.ca/eplant) (Toufighi et al. 2005). To the best of our knowledge, research has not been conducted so far on the effect of alteration in the function of this gene on plant phenotypes, but is known to be the target of a strong herbicide, hydantocydin (Siehl et al. 1996), which again highlights the central role of this enzyme in plant cell metabolism.
For female E479, 31 probe markers, from 29 different scaffolds, are positioned within a 5 cm distance from the Pendula marker, with two probe markers positioned at the same position as the Pendula marker (Supplementary Table S2). These two probe markers are located within the gene models MA_312116g0010, transcribing a lumazine-binding family protein involved in riboflavin synthase/biosynthesis, and MA_10432730g0010, transcribing a P-loop containing nucleoside triphosphate hydrolases superfamily protein. Both of these probe markers are present as double heterozygotes in the parents (E479 × E2089) but MA_312116:1 shows the most significant p-value of the double heterozygous markers for the cross (p-value 7.17e-30, Table 3). MA_312116:1 is also present as a double heterozygous marker in the E477 × K954 cross and there positioned 1.22 cm away from the Pendula marker in the E477 map and shows the strongest association of all double heterozygous markers (p-value 1.14e-31, (Supplementary Table S1)). The probe marker showing the strongest association for cross E479 × E2089 is MA_114136:1 (p-value 1.72e-61) located within the gene model MA_114136g0010, which transcribes a ribosomal protein S26e family protein and is positioned 0.48 cm away from the Pendula marker on the E479 map (Supplementary Table S2).
There are 12 probe markers significantly associated with the Pendula marker in both crosses (Table 2). Those with the highest p-values are MA_34514:1 and MA_10429386:1 located within the gene models MA_34514g0010 and MA_10429386g0010, respectively, which transcribe a membrane trafficking VPS54 family protein. Furthermore, non-synonymous SNPs were detected in the coding regions of five Norway spruce genes. Supplementary Table S3 presents the details of these genes including the alignment information of their corresponding proteins with Arabidopsis proteins (Supplementary Figs. 3–7) representing the actual alignments performed with MUSCLE (Edgar 2004). Four genes gave the corresponding BLAST hit in Arabidopsis (Supplementary Table S3). The putative Arabidopsis orthologues of these genes are involved in various plant processes such as growth, defense, and stress response. GLUTATHIONE S-TRANSFERASEs is a huge gene family. One of the members of this family, GLUTATHIONE S-TRANSFERASE U17 (ATGSTU17; AT1G10370) is involved in the modulation of seedling development in Arabidopsis. ATGSTU17 participates in the regulation of the architecture of Arabidopsis inflorescence by regulating the expression of AtMYB13 gene, which in turn acts at branching points of the inflorescence (Jiang et al. 2010; Kirik et al. 1998). No significant sequence similarity was detected between AT1G10370 and its ortholog in spruce MA_5480022p0010, yet the amino acid tryptophan (W) where the SNPs was located appears to be conserved between both species (Supplementary Figure S7). The only marker that presents common non-synonymous SNPs in both crosses is MA_51707:1. Its closest BLAST hit in Arabidopsis is AT4G04350—EMBRYO DEFECTIVE 2369 (EMB2369; tRNA synthetase class I family protein). This gene is highly expressed in young leaves and pedicels (Klepikova atlas). Aminoacyl-transfer RNA synthetases have been identified as key players in translation and they have an early role in protein synthesis (Brandao and Silva-Filho, 2011). It is involved in embryo and plant development (Meinke, 2020). One of the orthologs of this gene was found to be related to the architecture of the embryo and kernel size in maize (X. Li et al. 2022). However, no information regarding phenotype changes in other species is available.
Among the previously mentioned 12 probe markers linked to the Pendula phenotype in both crosses, only 5 markers shared the SNP position. Four of them were located outside the coding regions of the genes. In the case of MA_312116:1 marker (the one with the most significant p-value of the double heterozygous markers), it encodes a gene with a putative Arabidopsis orthologue (AT2G20690) with riboflavin synthesis enzymatic function (Fischer et al. 2005). Riboflavin synthesis has been proposed to affect to the cytokinin and auxin balance/signaling (Hedtke et al. 2012) and therefore leading to different branching phenotypes. For the marker MA_10426685:1, its closest Arabidopsis BLAST is AT1G74240. This gene encodes a mitocondrial transmembrane transport. However, this gene has not been linked with any function that might explain the studied phenotype. MA_51707:1 (previously described with its potential role in cytokinin and auxin synthesis/signaling). Closest Arabidopsis BLAST to the marker MA_881406:1 is AT1G10380, encoding for a putative membrane lipoprotein highly expressed in root apex (Klepikova atlas). Finally, MA_335371:1 putative Arabidopsis orthologue is AT1G04960, a protein of unknown function (DUF1664) expressed in senescent leaf petioles (Klepikova atlas). We propose that further research in the functions of the genes linked to markers MA_312116:1 and MA_51707:1 (MA_312116g0010 and MA_51707g0010, respectively) may give insights into tree architecture in Norway spruce, particularly about the Pendula branching phenotype.
Since the genetic maps only managed to anchor ~ 26% of all partially validated gene models in the P. abies genome assembly v 1.0 (Nystedt et al. 2013), there is a risk that we missed the causative gene. However, creating a genetic map that anchors all 66,632 validated gene models is not feasible until a less fragmented and more complete genome assembly with additional gene models per scaffold is made available. Once a better assembly of the genome is available, the generated data in this research should be reanalyzed to provide further insights into the genetic nature of the “Pendula” Norway spruce phenotype.
Data availability
Raw data is available at https://doi.org/10.5281/zenodo.7093290
References
Bateman A, Martin MJ, Orchard S, Magrane M, Agivetova R, Ahmad S, Alpi E, Bowler-Barnett EH, Britto R, Bursteinas B, Bye-A-Jee H, Coetzee R, Cukura A, da Silva A, Denny P, Dogan T, Ebenezer TG, Fan J, Castro LG., … Zhang J (2021) UniProt: The universal protein knowledgebase in 2021. Nucleic Acids Res, 49(D1), D480–D489. https://doi.org/10.1093/NAR/GKAA1100
Bemer M, van Mourik H, Muiño JM, Ferrándiz C, Kaufmann K, Angenent GC (2017) FRUITFULL controls SAUR10 expression and regulates Arabidopsis growth and architecture. J Exp Bot 68(13):3391. https://doi.org/10.1093/JXB/ERX184
Bernhardsson C, Vidalis A, Wang X, Scofield DG, Schiffthaler B, Baison J, Street NR, Rosario García-Gil M, Ingvarsson PK (2019) An ultra-dense haploid genetic map for evaluating the highly fragmented genome assembly of Norway spruce (Picea abies). G3 (Bethesda, Md.) 9(5):1623–1632
Cartwright DA, Troggio M, Velasco R, Gutin A (2007) Genetic mapping in the presence of genotyping errors. Genetics 176(4):2521–2527. https://doi.org/10.1534/GENETICS.106.063982
Danecek P, Auton A, Abecasis G, Albers CA, Banks E, DePristo MA, Handsaker RE, Lunter G, Marth GT, Sherry ST, McVean G, Durbin R (2011) The variant call format and VCFtools. Bioinformatics 27(15):2156–2158. https://doi.org/10.1093/bioinformatics/btr330
Dardick C, Callahan A, Horn R, Ruiz KB, Zhebentyayeva T, Hollender C, Whitaker M, Abbott A, Scorza R (2013) PpeTAC1 promotes the horizontal growth of branches in peach trees and is a member of a functionally conserved gene family found in diverse plants species. Plant J 75(4):618–630. https://doi.org/10.1111/tpj.12234
DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, Philippakis AA, del Angel G, Rivas MA, Hanna M, McKenna A, Fennell TJ, Kernytsky AM, Sivachenko AY, Cibulskis K, Gabriel SB, Altshuler D, Daly MJ (2011) A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet 43(5):491–498. https://doi.org/10.1038/ng.806
Edgar RC (2004) MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32(5):1792–1797. https://doi.org/10.1093/nar/gkh340
Fischer M, Haase I, Feicht R, Schramek N, Köhler P, Schieberle P, Bacher A (2005) Evolution of vitamin B2 biosynthesis: Riboflavin synthase of Arabidopsis thaliana and its inhibition by riboflavin. Biol Chem 386(5):417–428. https://doi.org/10.1515/BC.2005.050
Fladung M (2021) Targeted crispr/cas9-based knock-out of the rice orthologs tiller angle control 1 (Tac1) in poplar induces erect leaf habit and shoot growth. Forests 12(12):1615. https://doi.org/10.3390/F12121615
Gerendiain AZ, Peltola H, Pulkkinen P, Ikonen VP, Jaatinen R (2008) Differences in growth and wood properties between narrow and normal crowned types of Norway spruce grown at narrow spacing in Southern Finland. Silva Fennica, 42(3). https://doi.org/10.14214/sf.247
Hedtke B, Alawady A, Albacete A, Kobayashi K, Melzer M, Roitsch T, Masuda T, Grimm B (2012) Deficiency in riboflavin biosynthesis affects tetrapyrrole biosynthesis in etiolated Arabidopsis tissue. Plant Mol Biol 78(1–2):77–93. https://doi.org/10.1007/S11103-011-9846-1/METRICS
Hollender CA, Dardick C (2015) Molecular basis of angiosperm tree architecture. New Phytol 206(2):541–556. https://doi.org/10.1111/nph.13204
Hollender CA, Pascal T, Tabb A, Hadiarto T, Srinivasan C, Wang W, Liu Z, Scorza R, Dardick C (2018) Loss of a highly conserved sterile alpha motif domain gene (WEEP) results in pendulous branch growth in peach trees. Proc Nat Acad Sci USA 115(20):4690–4699. https://doi.org/10.1073/pnas.1704515115
Hollender CA, Waite JM, Tabb A, Raines D, Chinnithambi S, Dardick C (2018) Alteration of TAC1 expression in Prunus species leads to pleiotropic shoot phenotypes. Hortic Res 5(1):1–9. https://doi.org/10.1038/s41438-018-0034-1
Jiang HW, Liu MJ, Chen IC, Huang CH, Chao LY, Hsieh HL (2010) A glutathione s-transferase regulated by light and hormones participates in the modulation of arabidopsis seedling development. Plant Physiol 154(4):1646–1658. https://doi.org/10.1104/pp.110.159152
Jiao Z, Du H, Chen S, Huang W, Ge L (2021) LAZY gene family in plant gravitropism. Front Plant Sci 11:2096. https://doi.org/10.3389/FPLS.2020.606241/BIBTEX
Karki L, Tigerstedt PMA (1985) Definition and exploitation of forest tree ideotypes in Finland. In: Cannell M.G.R., Jackson J.E. (eds) Attributes of trees as crop plants. [Abbots Ripton, Huntingdon]: Institute of Terrestrial Ecology
Kelley D, Salzberg S (2010) Detection and correction of false segmental duplications caused by genome mis-assembly. Genome Biol 11(3):R28
Khan MA, Han Y, Zhao YF, Troggio M, Korban SS (2012) A multi-population consensus genetic map reveals inconsistent marker order among maps likely attributed to structural variations in the apple genome. PLOS ONE 7(11):e47864. https://doi.org/10.1371/JOURNAL.PONE.0047864
Kirik V, Kölle K, Wohlfarth T, Miséra S, Bäumlein H (1998) Ectopic expression of a novel MYB gene modifies the architecture of the Arabidopsis inflorescence. Plant J 13(6):729–742. https://doi.org/10.1046/j.1365-313X.1998.00072.x
Lehner A, Pöykkö T, Lehner A, Campbell M, Gliissl J, Kreike J, Neale DB, Wheeler NC (1995) Breeding for crop trees view project from sustainable design to sustainable implementation-knowledge value chains for green economy view project identification of a RAPD marker linked to the pendula gene in Norway spruce (Picea abies (L .) Karst. f. pendula). Theor Appl Genet 91:1092–1094. https://doi.org/10.1007/BF00223924
Lepistö M (1985) Riippakuusen periytymisestä ja kapealavaisuuden hyväksikäytöstä kuusen jalostukessa. Summary: The inheritance of pendula spruce (Picea abies f. pendula) and utilization of the narrow-crowned type in spruce breeding. Found For Tree Breding, Inf 1:1–6
Leyser O (2008) Strigolactones and shoot branching: A new trick for a young dog. Developmental Cell 15(3):337–338. https://doi.org/10.1016/j.devcel.2008.08.008
Li H, Durbin R (2009) Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25(14):1754–1760. https://doi.org/10.1093/bioinformatics/btp324
Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, 1000 Genome Project Data Processing Subgroup (2009) The sequence alignment/map format and SAMtools. Bioinformatics 25(16):2078–2079. https://doi.org/10.1093/bioinformatics/btp352
Li X, Wang M, Zhang R, Fang H, Fu X, Yang X, Li J (2022) Genetic architecture of embryo size and related traits in maize. Crop J 10(1):204–215. https://doi.org/10.1016/j.cj.2021.03.007
Marchler-Bauer A, Derbyshire MK, Gonzales NR, Lu S, Chitsaz F, Geer LY, Geer RC, He J, Gwadz M, Hurwitz DI, Lanczycki CJ, Lu F, Marchler GH, Song JS, Thanki N, Wang Z, Yamashita RA, Zhang D, Zheng C, Bryant SH (2015) CDD: NCBI’s conserved domain database. Nucleic Acids Res 43(Database issue):D222–D226. https://doi.org/10.1093/NAR/GKU1221
Margarido GRA, Souza AP, Garcia AAF (2007) OneMap: software for genetic mapping in outcrossing species. Hereditas 144(3):78–79. https://doi.org/10.1111/J.2007.0018-0661.02000.X
McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, DePristo MA (2010) The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 20(9):1297–1303. https://doi.org/10.1101/gr.107524.110
Nystedt B, Street NR, Wetterbom A, Zuccolo A, Lin YC, Scofield DG, Vezzi F, Delhomme N, Giacomello S, Alexeyenko A, Vicedomini R, Sahlin K, Sherwood E, Elfstrand M, Gramzow L, Holmberg K, Hällman J, Keech O, Klasson L., … Jansson S (2013) The Norway spruce genome sequence and conifer genome evolution. Nature 2013 497:7451, 497(7451), 579–584. https://doi.org/10.1038/nature12211
Pang YLJ, Poruri K, Martinis SA (2014) tRNA synthetase: TRNA aminoacylation and beyond. Wiley Interdiscip Rev: RNA 5(4):461–480. https://doi.org/10.1002/wrna.1224
Pöykkö VT, Pulkkinen PO (1990) Characteristics of normal-crowned and pendula spruce ( Picea abies (L.) Karst.) examined with reference to the definition of a crop tree ideotype. Tree Physiol 7(1):201–207. https://doi.org/10.1093/treephys/7.1-2-3-4.201
Pulkkinen P, Poykko T (1990) Inherited narrow crown form, harvest index and stem biomass production in Norway spruce, Picea abies. Tree Physiol 6(4):381–391. https://doi.org/10.1093/treephys/6.4.381
R core team (2013) R core team. In R: A Language and Environment for Statistical Computing (vol. 55)
Roychoudhry S, Kepinski S (2015) Shoot and root branch growth angle control-the wonderfulness of lateralness. Curr Opin Plant Biol 23(124):131. https://doi.org/10.1016/j.pbi.2014.12.004
Sakai T, Haga K (2012) Molecular genetic analysis of phototropism in arabidopsis. Plant Cell Physiol 53(9):1517–1534. https://doi.org/10.1093/PCP/PCS111
Salzberg S, Yorke J (2005) Beware of mis-assembled genomes. Bioinformatics 21(24):4320–4321
Sax H, Sax K (1933) Chromosome number and morphology in the conifers. J Arnold Arboretum 14:356–375
Schiffthaler B, Bernhardsson C, Ingvarsson PK, Street NR (2017) BatchMap: a parallel implementation of the OneMap R package for fast computation of F1 linkage maps in outcrossing species. PLOS ONE 12(12):e0189256. https://doi.org/10.1371/JOURNAL.PONE.0189256
Siehl DL, Subramanian MV, Walters EW, Lee S-F, Anderson RJ, Toschi AG (1996) Adenylosuccinate synthetase: site of action of hydantocidin, a microbial phytotoxin. Plant Physiol 110:753–758
Smith PMC, Atkins CA (2002) Purine biosynthesis. Big in cell division, even bigger in nitrogen assimilation. Plant Physiol 128(3):793–802. https://doi.org/10.1104/PP.010912
Stayton MM, Rudolph FB, Fromm HJ (1983) Regulation, genetics, and properties of adenylosuccinate synthetase: a review. Curr Top Cell Regul 22(C):103–141. https://doi.org/10.1016/B978-0-12-152822-5.50008-7
Strohm A, Baldwin K, Masson PH (2013) Gravitropism in Arabidopsis thaliana. Brenner’s Encyclopedia of Genetics, 2nd edition, 358–361. https://doi.org/10.1016/B978-0-12-374984-0.00662-8
Toufighi K, Brady SM, Austin R, Ly E, Provart NJ (2005) The Botany Array Resource: e-Northerns, expression angling, and promoter analyses. Plant J 43(1):153–163. https://doi.org/10.1111/J.1365-313X.2005.02437.X
Van der Auwera GA, Carneiro MO, Hartl C, Poplin R, del Angel G, Levy-Moonshine A, Jordan T, Shakir K, Roazen D, Thibault J, Banks E, Garimella KV, Altshuler D, Gabriel S, DePristo MA, Auwera GA, Carneiro MO, Hartl C, Poplin R., … DePristo MA (2013) From FastQ data to high-confidence variant calls: The genome analysis toolkit best practices pipeline. In Current Protocols in Bioinformatics (pp. 11.10.1–11.10.33). John Wiley & Sons, Inc. https://doi.org/10.1002/0471250953.bi1110s43
Vidalis A, Scofield DG, Neves LG, Bernhardsson C, García-Gil MR, Ingvarsson PK (2018) Design and evaluation of a large sequence-capture probe set and associated SNPs for diploid and haploid samples of Norway spruce (Picea abies). BioRxiv, 291716. https://doi.org/10.1101/291716
Wagner U, Edwards R, Dixon DP, Mauch F (2002) Probing the diversity of the Arabidopsis glutathione S-transferase gene family. Plant Mol Biol 49(5):515–532. https://doi.org/10.1023/A:1015557300450
Waite JM, Dardick C (2020) IGT/LAZY family genes are differentially influenced by light signals and collectively required for light-induced changes to branch angle. BioRxiv, 2020.07.15.205625. https://doi.org/10.1101/2020.07.15.205625
Xu D, Qi X, Li J, Han X, Wang J, Jiang Y, Tian Y, Wang Y (2017) PzTAC and PzLAZY from a narrow-crown poplar contribute to regulation of branch angles. Plant Physiol Biochem : PPB 118:571. https://doi.org/10.1016/J.PLAPHY.2017.07.011
Funding
Open access funding provided by Swedish University of Agricultural Sciences. This work was supported by grants from the Knut and Alice Wallenberg Foundation and the Swedish Governmental Agency for Innovation Systems (VINNOVA). CB and the project was funded by Kempestiftelserna foundation.
Author information
Authors and Affiliations
Contributions
RGG planned the project; POP organized the sample collection; CB extracted DNA; CB and DGS performed alignment and SNP calling; CB and PKI filtered the data and made all maps and QTL analysis; FGM and SSR performed deeper SNP analysis, gene alignments of candidate genes, and major modifications to the original draft; CB wrote the first draft; FGM and SRR finalized the draft; all authors commented to the final draft of the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Data archiving statement
The VCF files and the genetic maps generated from the genotyping of crossings are available at https://doi.org/10.5281/zenodo.7093290. Further information regarding sequences and alignments are included in supplementary files.
Additional information
Communicated by S.C. González-Martínez.
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Francisco Gil-Muñoz and Carolina Bernhardsson shared first authorship.
Supplementary information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Gil-Muñoz, F., Bernhardsson, C., Ranade, S.S. et al. QTL mapping of the narrow-branch “Pendula” phenotype in Norway spruce (Picea abies L. Karst.). Tree Genetics & Genomes 19, 28 (2023). https://doi.org/10.1007/s11295-023-01599-6
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11295-023-01599-6